Our Research
We study the ecological and evolutionary dynamics of infectious disease in animal and human populations, with emphasis on zoonotic and emerging pathogens. By combining theoretical models and data, we aim to deepen our understanding of fundamental principles of disease transmission and adaptation, and apply those principles to interpret observed patterns, uncover driving mechanisms, and design effective control policies.
Zoonoses -- i.e. diseases that transmit from animals to humans -- account for over 60% of all human pathogens and 75% of emerging pathogens, but key aspects of zoonotic disease dynamics remain poorly understood. We have projects focusing on each phase of the zoonotic emergence process:
- Dynamics of zoonotic infections in their wildlife reservoirs
- Spillover transmission from animals to humans
- Outbreak dynamics of newly introduced pathogens
- Adaptation of pathogens to new host species
This work spans a range of pathogen-host systems, including leptospirosis in California sea lions, monkeypox at the wildlife-human interface, and influenza and other emerging diseases in human populations. There are also several conceptual themes running throughout the work, including the impact of host heterogeneities and superspreading on disease dynamics and control, and the integration of mechanisms across disciplines and across scales of biological organization.
The need for integration
 Our recent review of all published models of zoonotic disease transmission published in Science showed that the field suffers from a marked lack of integration (see figure). Very few modeling studies bring together the animal and human components of zoonotic transmission, to focus on cross-species spillover transmission or the ‘stuttering chains’ of transmission that occur when a pathogen transmits weakly between humans. Models that integrate pathogen evolution alongside transmission dynamics have also been rare. In fact, most modeling studies have focused on a single phase of the process, missing out on the essential cross-species nature of zoonotic infections – this is especially true for directly-transmitted pathogens such as influenza or monkeypox. There has also been too little integration across the many disciplines that influence disease dynamics at individual, population and global scales.
Our recent review of all published models of zoonotic disease transmission published in Science showed that the field suffers from a marked lack of integration (see figure). Very few modeling studies bring together the animal and human components of zoonotic transmission, to focus on cross-species spillover transmission or the ‘stuttering chains’ of transmission that occur when a pathogen transmits weakly between humans. Models that integrate pathogen evolution alongside transmission dynamics have also been rare. In fact, most modeling studies have focused on a single phase of the process, missing out on the essential cross-species nature of zoonotic infections – this is especially true for directly-transmitted pathogens such as influenza or monkeypox. There has also been too little integration across the many disciplines that influence disease dynamics at individual, population and global scales.
Our lab’s work aims to address this gap in integrative research, by pursuing five major projects that overlap to span all phases of the zoonotic emergence process.
- Leptospirosis in California sea lions and other wildlife species
- Cross-scale models of zoonotic viruses (including SARS-CoV-2)
- Childhood hemagglutinin imprinting to influenza A viruses
- Monkeypox emergence in the Democratic Republic of Congo
- Cross-scale models for evolutionary emergence of novel pathogens
By working on these projects in parallel, we identify common themes in zoonotic disease dynamics and benefit from cross-pollination between systems, as ideas from one study help to solve problems in another.
Our Major Projects
Leptospirosis in California sea lions and other wildlife species
Leptospirosis is thought to be the most widespread zoonotic disease in the world, and it imposes a high health burden in humans, wildlife and livestock worldwide. Yet there are many open questions about how the bacteria that cause leptospirosis (pathogen Leptospira species) spread among animals in wildlife and livestock reservoir populations, and this limits our ability to understand risks to spillover hosts such as humans.
We are working to address this gap, by studying the factors that drive recurring, deadly leptospirosis outbreaks in California sea lions (Zalophus californianus) along the Pacific coast of the US.  Every fall, the disease causes sea lions to strand and die, and some years there are dramatic outbreaks that cause major mortality in the sea lion population. In collaboration with a broad team of partners, including scientists and veterinarians at The Marine Mammal Center and the National Marine Fisheries Service, we have assembled an extensive data set describing disease spread in this system for the past three decades. With support from NSF, we are studying the disease in field, lab and clinical settings, and working with top microbiologists to conduct molecular analyses of the pathogen. Our findings are shedding new light on how the disease circulates among sea lions, why it causes major outbreaks in some years, and how it has persisted in the ecosystem for decades. We are also gaining new insights into the epidemiology of leptospirosis in animal populations. We are also using the data set to address classical questions in population ecology, such as the balance of intrinsic and extrinsic drivers in generating observed population dynamics.
Every fall, the disease causes sea lions to strand and die, and some years there are dramatic outbreaks that cause major mortality in the sea lion population. In collaboration with a broad team of partners, including scientists and veterinarians at The Marine Mammal Center and the National Marine Fisheries Service, we have assembled an extensive data set describing disease spread in this system for the past three decades. With support from NSF, we are studying the disease in field, lab and clinical settings, and working with top microbiologists to conduct molecular analyses of the pathogen. Our findings are shedding new light on how the disease circulates among sea lions, why it causes major outbreaks in some years, and how it has persisted in the ecosystem for decades. We are also gaining new insights into the epidemiology of leptospirosis in animal populations. We are also using the data set to address classical questions in population ecology, such as the balance of intrinsic and extrinsic drivers in generating observed population dynamics.
We have recently expanded this project to study leptospirosis in some terrestrial mammals in the California coastal ecosystem, including the endangered Channel Island fox (Urocyon littoralis). This project, which is a collaboration with the National Park Service, will explore the community ecology of disease spread in this system, and the possibility of marine-terrestrial transmission.
Cross-scale models of zoonotic viruses (including SARS-CoV-2)
In the context of the COVID-19 pandemic, we draw upon our existing research on novel bat viruses and human respiratory pathogens to contribute to the ongoing effort to understand and combat the SARS-CoV-2 virus. With the BatOneHealth team, we have been conducting research on henipaviruses (e.g. Nipah, Hendra) at the bat-human interface. We draw upon this prior work and the network of collaborators we had developed through it to address SARS-CoV-2, a bat coronavirus. We develop a variety of projects that use theoretical and mathematical models to extract better information from experimental and epidemiological data, linking within-host processes to population scale dynamics.
![]()
- In collaboration with colleagues at the NIH Rocky Mountain Laboratories (RML) (Munster Lab), we study the persistence of the virus in aerosols and on surfaces, developed mechanistic models of virus persistence as a function of temperature and humidity, and use this knowledge to guide pandemic response.
- We develop kinetic models of individual SARS-CoV-2 infections, which we used to study dose-dependent disease progression in animal model data generated by collaborators at NIH RML (Munster Lab) and Cornell University (Aguilar Lab).
- Using quantitative models of the immune response, we develop tools and best practice guidance for using serological data to characterize SARS-CoV-2 epidemiology.
Childhood hemagglutinin imprinting to influenza A viruses
Influenza pandemics occur every few decades, and persistently threaten to cause massive numbers of infections and fatalities, and halt the global economy. Pandemics occur when unfamiliar influenza viruses jump from animals into humans, and then manage to spread around the globe. Our lab is currently undertaking work to help forecast which age groups would be at the highest risk of severe infections in future pandemics, and which zoonotic influenza viruses would face the most resistance from pre-existing immunity if they were ever to jump from animals into humans.
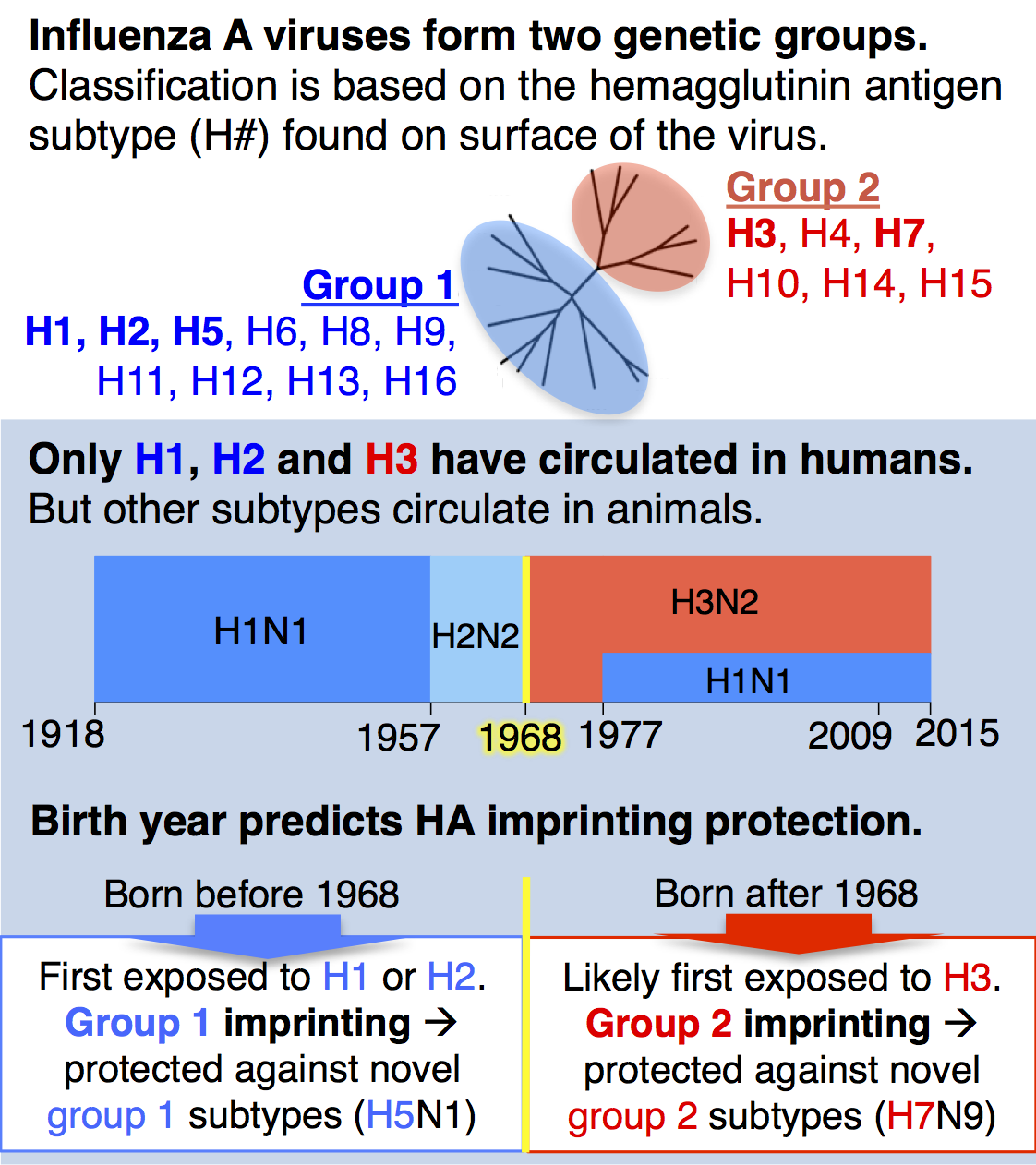
Until recently, a key idea in influenza epidemiology was that the entire human population would lack immunity against an animal-origin pandemic virus. However, our recent study with collaborator Michael Worobey showed for the first time that the human population actually has strong, pre-existing immunity against novel, zoonotic influenza A viruses with pandemic potential, and that this protection is predictably distributed across birth years. We analyzed data on all known cases of H5N1 and H7N9, two avian influenza viruses of great concern for pandemic emergence in humans, and showed that individuals gain lifelong, partial protection against novel hemagglutinin (HA) subtypes in the same genetic group as the influenza virus that caused their first infection during childhood (see diagram). We call this phenomenon, childhood HA imprinting.
As a birth year, 1968 marks a crucial turning point in HA imprinting protection. Anyone born before 1968 would have imprinted to HA group 1 (H1 or H2), whereas those born after 1968 have a much higher probability of imprinting to HA group 2 (H3, see figure at left). HA imprinting patterns explain why the majority of H5N1 cases observed to date have affected children and younger adults, while H7N9 has disproportionately affected older adults.
We can use routinely collected influenza surveillance data and demographic information to estimate the probability that an individual born in a particular year was first exposed to an influenza virus in group 1 or group 2. With this information, we are building models to forecast age distributions of infection in future pandemics, and to forecast whether potential pandemic viruses from group 1 or group 2 would face more resistance from pre-existing human immunity. We are also working with empirical collaborators to identify the specific immune mechanism responsible for HA imprinting protection. Additionally, we are working to understand whether vaccination in children has a negative or positive effect on HA imprinting protection, and to understand the impact of HA imprinting on the epidemiology of seasonal influenza viruses H1N1 and H3N2.
Monkeypox emergence in the Democratic Republic of Congo
The greatest known threat of new human pathogens comes from 'stuttering' zoonoses. These are pathogens that exhibit limited transmission among humans—enough to cause a cluster of cases, but not enough to trigger an epidemic. Such stuttering chains of transmission are red flags for emergence risk, since the barrier of human-to-human transmission has been breached, but they have proven very difficult to study due to their rare and transient nature. Methods to analyze epidemiological data from these pathogens are sorely lacking (see figure from Science paper, above).
 With Anne Rimoin in the UCLA School of Public Health and a team of collaborators, we are studying the dynamics of monkeypox at the wildlife-human interface in the Democratic Republic of the Congo. Monkeypox is a perfect case study for viral emergence: it transmits inefficiently among humans, but its transmission is rising as population immunity against poxviruses drops with time since the eradication of smallpox. We have extensive data on monkeypox epidemiology in the Congo basin from a historic surveillance program in the 1980s, and from on-going surveillance led by the UCLA team. This presents a unique opportunity to develop and test new models against two data sets that differ systematically in the degree of transmission
With Anne Rimoin in the UCLA School of Public Health and a team of collaborators, we are studying the dynamics of monkeypox at the wildlife-human interface in the Democratic Republic of the Congo. Monkeypox is a perfect case study for viral emergence: it transmits inefficiently among humans, but its transmission is rising as population immunity against poxviruses drops with time since the eradication of smallpox. We have extensive data on monkeypox epidemiology in the Congo basin from a historic surveillance program in the 1980s, and from on-going surveillance led by the UCLA team. This presents a unique opportunity to develop and test new models against two data sets that differ systematically in the degree of transmission
Our group is developing new modeling approaches to understand the rise of monkeypox in the Congo basin, and building tools to analyze data from any stuttering zoonosis. These include methods for assessing whether cases were infected by animal or human sources, and methods to estimate transmission parameters from the size distribution of human outbreaks. We are integrating these tools with disease transmission models to study the drivers of temporal and spatial patterns in monkeypox incidence, and to answer crucial questions such as how efficiently the virus spreads between humans. We’re using ideas from community ecology to study how smallpox eradication has altered monkeypox epidemiology, to find general lessons for current and future disease eradication campaigns. In addition, the tools we’re building will be applicable to other zoonotic threats such as H5N1 avian influenza or to re-emerging infections such as measles or pertussis in populations with low vaccine uptake.
Cross-scale models for evolutionary emergence of novel pathogens
Adaptive evolution can play a powerful role in pathogen emergence across species boundaries. A fast-growing empirical literature, propelled by sequencing technology, reverse genetics, and experimental infection studies, is mapping the impact of mutations on viral fitness across host species. But there is no framework to synthesize these findings in light of the key biological mechanisms at play, from viral growth within host individuals to the epidemiological processes of spillover, stuttering chains, and emergence.
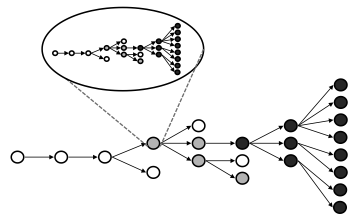 In an NSF-funded project in collaboration with Sebastian Schreiber at UC Davis we are working to construct a multi-scale theory for pathogen emergence, combining concepts from virology, population genetics, and epidemic modeling (see schematic figure). We are building from the smallest scales up, studying how basic processes of viral replication and within-host pathogen dynamics can influence processes at larger scales. So far we have studied how evolutionary emergence processes within hosts are impacted by mechanisms of viral genome replication, and how the cost of deleterious mutations balances with the benefit of adaptive evolution to influence emergence probability. We are constructing a series of cross-scale models that link within-host selection to evolutionary emergence that can occur in the course of a stuttering chain of transmission.
In an NSF-funded project in collaboration with Sebastian Schreiber at UC Davis we are working to construct a multi-scale theory for pathogen emergence, combining concepts from virology, population genetics, and epidemic modeling (see schematic figure). We are building from the smallest scales up, studying how basic processes of viral replication and within-host pathogen dynamics can influence processes at larger scales. So far we have studied how evolutionary emergence processes within hosts are impacted by mechanisms of viral genome replication, and how the cost of deleterious mutations balances with the benefit of adaptive evolution to influence emergence probability. We are constructing a series of cross-scale models that link within-host selection to evolutionary emergence that can occur in the course of a stuttering chain of transmission.
Ultimately, this research aims to connect laboratory results in molecular virology to sequence data collected in experimental infections and field surveillance, to guide rational assessment of the risk from possible zoonotic threats. With these goals in mind, we are continuing to develop our models to incorporate greater realism and make connections to current empirical research. In collaboration with Ren Sun in the UCLA medical school, we are using our models to analyze data from high-throughput experiments characterizing fitness and drug resistance phenotypes of viruses such as hepatitis C virus.
A related theme explores the role of defective virus particles in viral evolution and emergence. This work has its roots in an exploration of the potential to engineer defective particles into ‘therapeutic interfering particles’ to combat HIV. This proposed approach is essentially a gene therapy that can replicate within a host if that host is infected with HIV. We conducted cross-scale modeling analyses of the epidemiological impact of this therapy, which showed its remarkable potential to target disease superspreaders autonomously. In a follow-up model of the evolutionary dynamics of these therapies, we show their potential to co-evolve with HIV and circumvent the problem of resistance evolution. We are now applying these concepts to observed data, studying a defective dengue virus that spread in Southeast Asia in 2001-03. Combining sequence data with epidemic models, we are studying the mechanisms that allow a defective virus to spread sustainably, and its impact on the epidemiology of wild-type dengue virus. These studies of defective particles are revealing new principles about the mechanisms of viral emergence, including the unforeseen potential for viral complementation help viral lineages cross fitness valleys. They are also motivating us to develop new methods to measure selection across scales from viral sequence data, which will contribute directly to current challenges in zoonotic emergence.